For the treatment of adult patients with moderately to severely active CD
STELARA® HAS A PROVEN SAFETY PROFILE THROUGH 5 YEARS (FROM PHASE 3 STUDIES AND OPEN-LABEL LTE)1,2
STELARA® HAS A PROVEN SAFETY PROFILE THROUGH 5 YEARS (FROM PHASE 3 STUDIES AND OPEN-LABEL LTE)1,2
The safety of STELARA® was evaluated in 1,407 patients with moderately to severely active CD (CDAI ≥220 and ≤450) in 3 randomized, double-blind, placebo-controlled, parallel-group, multicenter studies through 1 year.
The overall safety profile of STELARA® in CD through 1 year was consistent with that seen in other approved indications.1
Common Adverse Reactions Through Week 8 in 2 Single IV Induction Studies
≥3% of patients treated with STELARA® and higher than placebo
STELARA®6 mg/kg IV single dose (N=470)
Placebo (N=466)
Vomiting
4%
3%
Other less common adverse reactions reported in patients in the 2 single IV induction studies included asthenia (1% vs 0.4%), acne (1% vs 0.4%), and pruritus (2% vs 0.4%).
Common Adverse Reactions Through Week 44 in SubQ Maintenance Study*
≥3% of patients treated with STELARA® and higher than placebo
STELARA®90 mg subQ every 8 weeks(N=131)
Placebo (N=133)†
Nasopharyngitis
11%
8%
Injection-site erythema
5%
0
Vulvovaginal candidiasis/mycotic infection
5%
1%
Bronchitis
5%
3%
Pruritus
4%
2%
Urinary tract infection
4%
2%
Sinusitis
3%
2%
Adverse Events THROUGH 1 Year1,2
Patients treated with STELARA®§ 90 mg subQ every 8 weeks
Placebo||
N (treated patients)‡
131
133
Average duration of follow-up (weeks)
35.2
32.0
Deaths
0
0
Percentage of patients
Adverse events
81.7%
83.5%
Serious adverse events
9.9%
15.0%
Infections
48.1%
49.6%
Serious infections
2.3%
2.3%
ADDITIONAL SAFETY AT 1 YEAR
<1% Hypersensitivity reactions1
One patient (0.08%) experienced signs and symptoms consistent with or related to a hypersensitivity reaction¶ after receiving the initial STELARA® IV dose. One patient (0.1%) experienced signs and symptoms consistent with anaphylaxis# after receiving a single subQ STELARA® dose. In both cases, patients were treated with oral antihistamines or corticosteroids and symptoms were resolved within an hour
<1% Malignancy rate**
Nonmelanoma skin cancer developed in 0.2% of both the STELARA® group and the placebo group
Other malignancies occurred in 0.2% of patients receiving STELARA® and in none of the patients receiving placebo
STELARA® is an immunosuppressant and may increase the risk of malignancy.
<3% Serious infections2
2.3% of patients treated with STELARA® 90 mg subQ every 8 weeks
2.3% of patients treated with placebo
<3% of patients developed antibodies1,2
2.9% of overall population treated with STELARA® and 2.4% of randomized patients treated with STELARA® 90 mg subQ every 8 weeks developed antibodies to the drug
*Week 44 was equivalent to 1 year after induction dose.1
†Patients who were in clinical response to STELARA® IV induction dosing and were randomized to placebo subQ on entry into this maintenance study.
‡Randomized patients in the maintenance study.1
§Included responders and nonresponders for STELARA® IV induction dosing.2
ǁIncludes: 1) All data for patients who were in clinical response to placebo IV induction dosing and received placebo subQ in the maintenance study. 2) The data from Week 8 onward for patients who were in clinical response to STELARA® IV induction dosing and received placebo subQ in this maintenance study.2
¶Symptoms included chest discomfort, flushing, urticaria, and increased body temperature.1
#Symptoms included tightness of the throat, shortness of breath, and flushing.1
**Malignancy rates included here reflect a combined evaluation of patients treated with STELARA® including those randomized to either the q8w or q12w dosing regimens.1,2
STELARA® Safety Through 1 Year in Combined CD+UC1,2*
ADVERSE EVENTS THROUGH 1 YEAR
Combined Safety (CD+UC)*CDUC
STELARA® 90 mg subQ every 8 weeks
Placebo†
STELARA®‡ 90 mg subQ every 8 weeks
Placebo§
STELARA® 90 mg subQ every 8 weeks
Placebo
N (treated patients)||
307
308
131
133
176
175
Average duration of follow-up (weeks)
39.2
37.8
35.2
32.0
42.2
42.3
Deaths
0
0
0
0
0
0
Percentage of patients
Adverse events
79.2%
80.8%
81.7%
83.5%
77.3%
78.9%
Serious adverse events
9.1%
12.0%
9.9%
15.0%
8.5%
9.7%
Infections
48.5%
47.7%
48.1%
49.6%
48.9%
46.3%
Serious infections
2.0%
2.3%
2.3%
2.3%
1.7%
2.3%
Combined Safety (CD+UC)*
STELARA® 90 mg subQ every 8 weeks
Placebo†
N (treated patients)||
307
308
Average duration of follow-up (weeks)
39.2
37.8
Deaths
0
0
Percentage of patients
Adverse events
79.2%
80.8%
Serious adverse events
9.1%
12.0%
Infections
48.5%
47.7%
Serious infections
2.0%
2.3%
CD
STELARA®‡ 90 mg subQ every 8 weeks
Placebo§
N (treated patients)||
131
133
Average duration of follow-up (weeks)
35.2
32.0
Deaths
0
0
Percentage of patients
Adverse events
81.7%
83.5%
Serious adverse events
9.9%
15.0%
Infections
48.1%
49.6%
Serious infections
2.3%
2.3%
UC
STELARA® 90 mg subQ every 8 weeks
Placebo
N (treated patients)||
176
175
Average duration of follow-up (weeks)
42.2
42.3
Deaths
0
0
Percentage of patients
Adverse events
77.3%
78.9%
Serious adverse events
8.5%
9.7%
Infections
48.9%
46.3%
Serious infections
1.7%
2.3%
*Data for combined Crohn’s disease and ulcerative colitis results include Phase 3 maintenance studies (Ulcerative Colitis: CNTO1275UCO3001; Crohn’s Disease: CNTO1275CRD3003); includes data up to the time of meeting loss of response criteria for patients who had Crohn’s disease.
ADDITIONAL SAFETY AT 1 YEAR FOR CD AND UC1
<1% Hypersensitivity reactions
In CD: Following initial STELARA® IV dose: 1 patient (0.08%) experienced signs and symptoms consistent with or related to a hypersensitivity reaction¶; following single subQ STELARA® dose: 1 patient (0.1%) experienced signs and symptoms consistent with anaphylaxis.# In both cases, patients were treated with oral antihistamines or corticosteroids and symptoms resolved within an hour.
No cases of anaphylactic or delayed hypersensitivity reactions with STELARA® through 1 year in the UC trials.
<1% Malignancy rates**
In CD: Nonmelanoma skin cancer developed in 0.2% of both the STELARA® group and the placebo group; other malignancies occurred in 0.2% of patients receiving STELARA® and in no patients receiving placebo.
In UC: Nonmelanoma skin cancer: STELARA®: 0.4%; placebo: 0.0%; other malignancies: STELARA®: 0.5%; placebo: 0.2%. STELARA® is an immunosuppressant and may increase the risk of malignancy.
≈2% Serious infections
In CD: 2.3% of patients in both the STELARA® 90 mg subQ every-8-weeks group and placebo group.
In UC: 1.7% of patients in the STELARA® 90 mg subQ every-8-weeks group reported serious infections vs 2.3% in the placebo group during the Phase 3 Maintenance Study.
<5% of patients developed antibodies
In CD: 2.9% of overall population treated with STELARA®; 2.4% of randomized patients treated with STELARA® 90 mg subQ every 8 weeks.
In UC: 4.6% of overall population treated with STELARA®; 3.4% of randomized patients treated with STELARA® 90 mg subQ every 8 weeks.
STELARA® COMBINED SAFETY PROFILE IN CD AND UC
Get details about the combined safety analysis of STELARA® as an induction and maintenance therapy for the treatment of adults with moderately to severely active CD and UC. The combined safety analysis pooled data are from six Phase 2 and Phase 3 CD and UC studies through 1 year of treatment, as published in Inflammatory Bowel Disease.
†Patients who were in clinical response to STELARA® IV induction dosing and were randomized to placebo subQ on entry into this maintenance study.
‡Included responders and nonresponders for STELARA® IV induction dosing.
§Includes: 1) All data for patients who were in clinical response to placebo IV induction dosing and received placebo subQ in the maintenance study. 2) The data from Week 8 onward for patients who were in clinical response to STELARA® IV induction dosing and received placebo subQ in this maintenance study.
||Randomized patients in the maintenance study.
¶Symptoms included chest discomfort, flushing, urticaria, and increased body temperature.
#Symptoms included tightness of the throat, shortness of breath, and flushing.
**Malignancy rates included here reflect a combined evaluation of patients treated with STELARA® including those randomized to either the q8w or q12w dosing regimens.
ADVERSE EVENTS THROUGH UP TO 5 YEARS IN CD2
NUMBER OF EVENTS PER 100 PATIENT-YEARS OF FOLLOW-UP (95% CI)*
STELARA®† 90 mg subQ every 8 weeks
Placebo‡
N (patients treated)
607
593
Average duration of follow-up (weeks)
122.55
37
Total patient years of follow-up
1431
422
Deaths
0.14 (0.02, 0.51)
0 (0.00, 0.71)
Adverse events
394.18 (383.96, 404.60)
545.86 (523.79, 568.62)
Serious adverse events
21.18 (19.46, 24.37)
29.15 (24.23, 34.78)
Infections§
99.19 (94.10, 104.49)
118.51 (108.35, 129.37)
Serious infections§
3.50 (2.59, 4.61)
6.16 (4.03, 9.03)
Data are presented by event rates adjusted by 100 patient-years to normalize difference in follow-up time between STELARA® and placebo treatment groups.
ADDITIONAL SAFETY THROUGH UP TO 5 YEARS2
0.99 Malignancy per 100 patient-years2II¶
STELARA® 90 mg every 8 weeks: 0.99 (n=354)#
Placebo: 1.70 (n=151)**
Patient-years of follow-up from Week 44 through the final safety visit for STELARA® (1110) and placebo (177)
<4 Serious infections per 100 patient-years2§II
STELARA® 90 mg every 8 weeks: 3.50†
Placebo: 6.16‡
<5%of patients developed antibodies to STELARA®2
Antibody formation among randomized patients who entered open-label LTE:
4.7% (50/1073) of all patients treated with at least 1 dose of STELARA® and 2.9% (3/102) of patients randomized to STELARA® 90 mg subQ every 8 weeks developed antibodies to the drug
IIData are presented by event rates adjusted by 100 patient-years to normalize difference in follow-up time between STELARA® and placebo treatment groups.
*Confidence intervals based on an exact method assuming that the observed number of events follows a Poisson distribution.
†Includes data from Week 0 of induction onward for patients who were rerandomized to ustekinumab 90 mg subQ q8w maintenance or patients who were not in clinical response to ustekinumab IV at Week 8 of the induction studies and received ustekinumab 90 mg subQ on entry into the maintenance study.
‡Includes data from Week 0 of induction onward up to the first ustekinumab dose for patients who were initially treated with placebo IV; includes data at or after 16 weeks from the first ustekinumab IV onward, up to the dose adjustment if patients had a dose adjustment, for patients who were rerandomized to placebo maintenance.
§Infections as assessed by the investigator.
¶Malignancy data are between Week 44 and Year 5 for patients who entered LTE.
#Includes: Patients who were in clinical response to ustekinumb IV induction dosing, randomized on entry into the maintenance study, received ustekinumab 90 mg subQ q8w, or met loss of response criteria from Week 8 through Week 32 and received ustekinumab 90 mg subQ q8w thereafter; patients who were not in clinical response to ustekinumab IV induction dosing, received ustekinumab 90 mg subQ at Week 0, achieved clinical response at Week 8, and initiated ustekinumab 90 mg subQ q8w.
**Includes: Patients who were in clinical response to ustekinumab IV induction dosing and received placebo subQ on entry into the maintenance study did not meet loss of response criteria from Week 8 through Week 32; patients who were in clinical response to placebo IV induction dosing and received placebo subQ on entry into the maintenance study.
References: 1. STELARA® [Prescribing Information]. Horsham, PA: Janssen Biotech, Inc. 2. Data on file. Janssen Biotech, Inc.
This publication includes information about STELARA® that is not found in the full Prescribing Information.
STELARA® (ustekinumab) is indicated for the treatment of adult patients with moderately to severely active Crohn’s disease who have:
failed or were intolerant to treatment with immunomodulators or corticosteroids, but never failed treatment with a tumor necrosis factor (TNF) blocker, or
failed or were intolerant to treatment with one or more TNF blockers.
Induction dose: A single IV infusion using weight-based dosage regimen: 260 mg (weight 55 kg or less), 390 mg (weight more than 55 kg to 85 kg), or 520 mg (weight more than 85 kg). Maintenance dose: A subQ 90-mg dose administered every 8 weeks after the induction dose. STELARA® can be administered by a healthcare provider or by the patient after physician approval and proper training.1
The article may only be available to subscribers of The New England Journal of Medicine. To access this content, you may have to sign in using your personal or institutional credentials. If you are not registered or a member of NEJM, then you may need to purchase this article through their site.
Do you want to leave STELARAhcp.com to access this clinical reprint?
STELARA®: ADVERSE EVENTS OF INTEREST AT YEAR 1 AND YEAR 3
At 1 year for patients treated with STELARA® in the maintenance study† (q8w 90 mg)
At 3 years for patients treated with STELARA® in the open-label long-term extension‡ (q8w 90 mg)
N (treated patients)
1,157
567
Adverse Events
75.1%
94.7%
Serious Adverse Events
14.7%
27.3%
Infections
42.2%
77.4%
Serious Infections
3.2%
10.4%
STELARA®: RATES OF ANTIBODY FORMATION
At 1 year among randomized patients treated with STELARA® in the maintenance study (q8w 90 mg)
At 3 years among randomized patients treated with STELARA® in the open-label long-term extension (q8w 90 mg)
N (treated patients)
127
82
Rates of Antibody Formation
2.4%
2.4%
ADDITIONAL SAFETY DURING MAINTENANCE STUDY (FROM WEEK 0 TO WEEK 44)
<1% Hypersensitivity Reactions1
One patient (0.08%) experienced signs and symptoms consistent with or related to a hypersensitivity reaction§ after receiving the initial STELARA® IV dose. One patient (0.1%) experienced signs and symptoms consistent with anaphylaxis|| after receiving a single subQ STELARA® dose. In both cases, patients were treated with oral antihistamines or corticosteroids and symptoms were resolved within an hour
<1% Malignancy Rate1
With up to 1 year of treatment, nonmelanoma skin cancer developed in 0.2% of both the STELARA® group and the placebo group. Other malignancies occurred in 0.2% of patients receiving STELARA® and in none of the patients receiving placebo. STELARA® is an immunosuppressant and may increase the risk of malignancy
ADDITIONAL SAFETY DURING OPEN-LABEL LTE (FROM WEEK 44 TO WEEK 156)
Treatment-emergent Malignancies2
STELARA® 90 mg every 8 weeks: 1.1% (4/354)
STELARA® 90 mg every 12 weeks: 1.9% (4/213)
Placebo: 2.0% (3/151)
Serious Infections2
STELARA®: 7.4% (42/567)
Placebo: 4.0% (6/151)
*156-week data (164 weeks after induction dose) included randomized and nonrandomized patients who entered the open-label LTE.2
†Included responders and nonresponders to STELARA® IV induction dosing.2
‡All randomized and nonrandomized patients treated with STELARA® who entered the open-label LTE.1
§Symptoms included chest discomfort, flushing, urticaria, and increased body temperature.1
||Symptoms included tightness of the throat, shortness of breath, and flushing.1
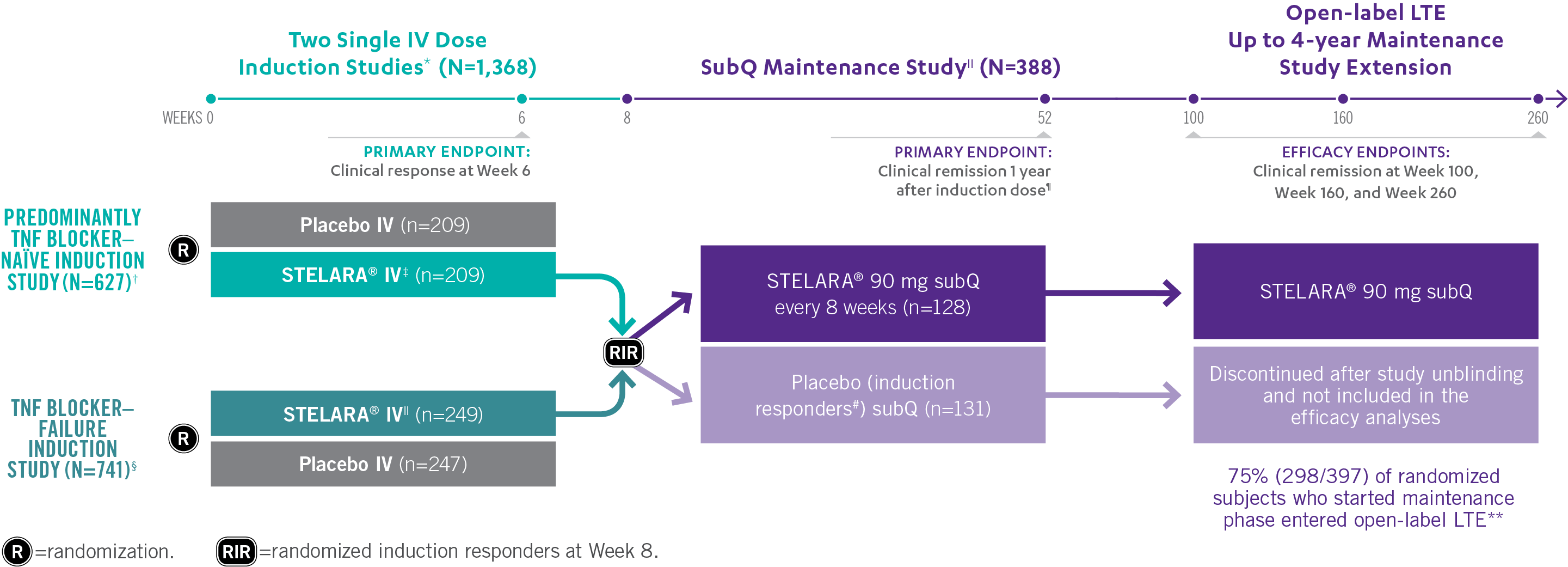
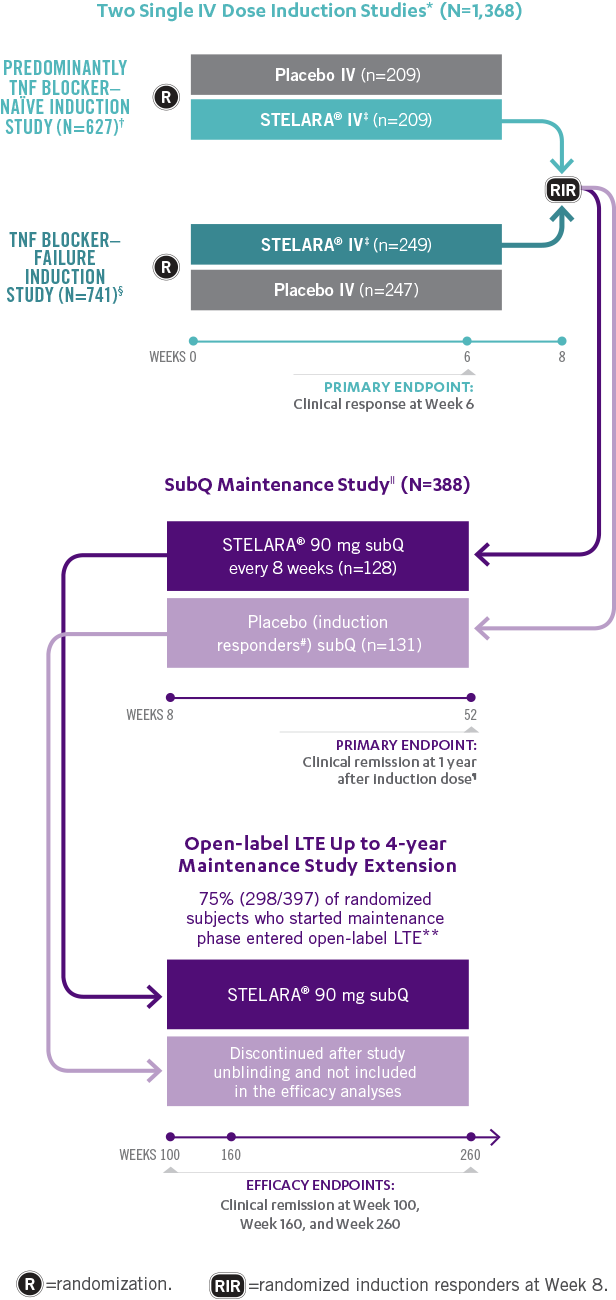
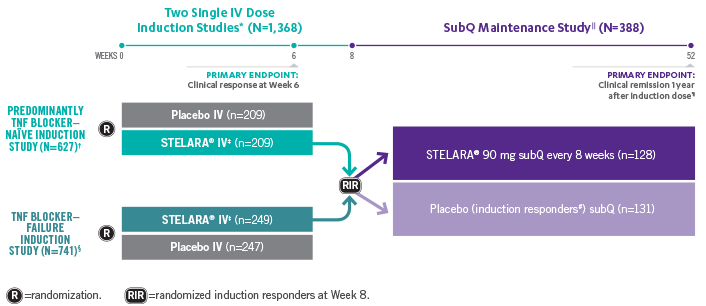
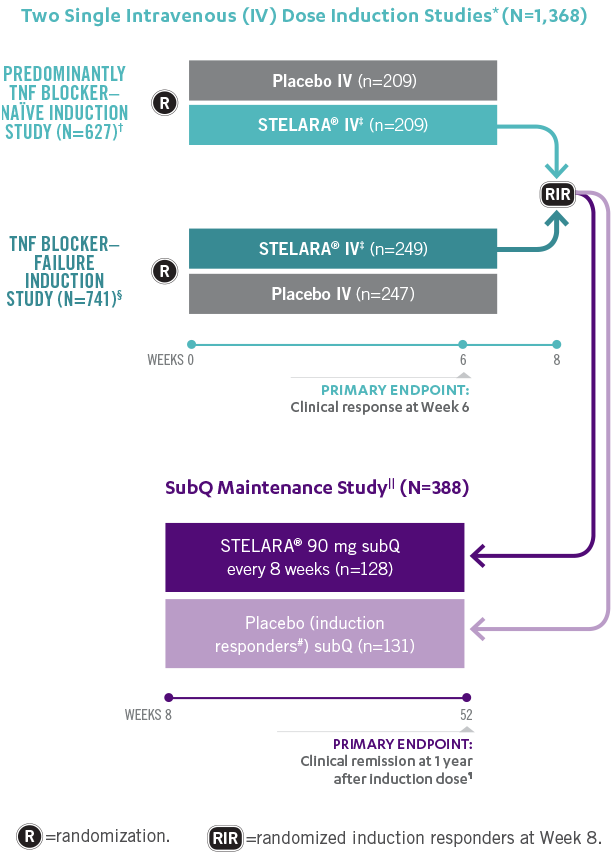
Studies THAT ASSESSED Efficacy and Safety From Week 3 to ≈5 YEARS1,2
*In both induction studies, patients were randomized to receive STELARA® weight-based dosage regimen (approximately 6 mg/kg), STELARA® 130-mg IV, or IV placebo. The 130-mg IV dose is not an approved induction dose and therefore is not shown. Clinical response was defined as reduction in CDAI score of ≥100 points or CDAI score of <150.1
†69% of patients were TNF blocker naïve. Remaining population were patients previously exposed to, but who did not fail, treatment with TNF blockers. All patients in the study failed or were intolerant to conventional treatment (eg, azathioprine, 6-mercaptopurine, methotrexate, or corticosteroids).1
‡Weight-based induction dosage regimen: STELARA® 260 mg (weight ≤55 kg), STELARA® 390 mg (weight >55 kg to 85 kg), and STELARA® 520 mg (weight >85 kg).1
§Patients were intolerant to or failed TNF blocker therapy.1
||The maintenance study included a third randomized arm where patients received STELARA® 90 mg subQ every 12 weeks. STELARA® 90 mg subQ every 12 weeks is not an approved maintenance dose and therefore is not shown. Patients randomized in the maintenance study were those who had a clinical response to STELARA® IV at Week 8 during either induction study.1,2
¶Clinical remission was defined as a CDAI score of <150. Week 44 of the maintenance study was defined as 1 year from initiation of the induction dose (8-week induction + 44-week maintenance study=1 year).1
#All patients randomized to placebo in the maintenance study had a single STELARA® IV induction dose.1
**Patients in the LTE study included those from the nonapproved STELARA® 90 mg subQ every-12-week maintenance arm, which is not represented in the figure above.2
STELARA® Clinical Trial Design: 3 Pivotal Studies With a Combined 1 Year1,2
*In both induction studies, patients were randomized to receive STELARA® weight-based dosage regimen (approximately 6 mg/kg), STELARA® 130-mg IV dose, or IV placebo. The 130-mg IV dose is not an approved induction dose and therefore is not shown. Clinical response was defined as reduction in CDAI score of ≥100 points or CDAI score of <150.1
†69% of patients were TNF blocker naïve. Remaining population was patients previously exposed to, but who did not fail, treatment with TNF blockers. All patients in the study failed or were intolerant to conventional treatment (eg, azathioprine, 6-mercaptopurine, methotrexate, or corticosteroids).1,2
§Patients were intolerant to or failed TNF blocker therapy.1
||The maintenance study included a third randomized arm where patients received STELARA® subQ 90 mg every 12 weeks. STELARA® 90 mg subQ every 12 weeks is not an approved maintenance dose.1,2
¶Clinical remission was defined as a CDAI score of <150.1
#All patients randomized to placebo in the maintenance study had a single STELARA® IV induction dose.1
This publication contains information about STELARA® that is not included in the full Prescribing Information.
Please see the full Prescribing Information for STELARA® for adequate directions for use for the approved indications.
STELARA® (ustekinumab) is indicated for the treatment of adult patients with moderately to severely active Crohn’s disease.
STELARA® (ustekinumab) is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis.
STELARA® DOSING INFORMATION FOR CROHN'S DISEASE AND ULCERATIVE COLITIS
Induction Dose: A single intravenous infusion using a weight-based dosage regimen: 260 mg (weight 55 kg or less), 390 mg (weight more than 55 kg to 85 kg), or 520 mg (weight more than 85 kg).
Maintenance Dose: A subcutaneous 90 mg dose administered 8 weeks after the initial intravenous dose, then every 8 weeks thereafter.
STELARA® is intended for use under the guidance and supervision of a physician with patients who will be closely monitored and have regular follow-up.
Do you want to leave STELARAHCP.com to access the clinical reprint?
STELARA® open-label LTE study design in adult patients with moderately to severely active UC
UNIFI CLINICAL TRIAL DESIGN
STUDY DESIGN IN UC1
Designed to prospectively evaluate clinical remission, clinical response, steroid-free clinical remission, and, for the first time in Phase 3 clinical trials, Histo-Endoscopic Mucosal Improvement (HEMI)
STELARA® CLINICAL TRIAL DESIGN: PHASE 3 STUDIES AND OPEN-LABEL LTE IN UC1,2
INDUCTION STUDY
MAJOR SECONDARY ENDPOINTS INCLUDED:
Clinical response at Week 8#
Endoscopic improvement at Week 8**
MAINTENANCE STUDY
MAJOR SECONDARY ENDPOINTS INCLUDED:
Maintenance of clinical response at 1 year after induction doseƗƗ
Endoscopic improvement at 1 year after induction dose**
Steroid-free clinical remission at 1 year after induction dose‡‡
Maintenance of clinical remission at 1 year§§
OPEN-LABEL LTE
All patients completing the Maintenance Study were eligible to enter the LTE. Placebo subQ patients were discontinued after unblinding at 1 year after induction dose. Efficacy measures were collected every 12 weeks thereafter. Safety was evaluated throughout.
OTHER SECONDARY ENDPOINTS IN THE INDUCTION AND MAINTENANCE STUDIES INCLUDED:
HEMI at Week 8 (other secondary endpoint) and at 1 year (other endpoint) after the induction doseIIII
Adult patients with moderately to severely active UC who had an inadequate response to or failed to tolerate biologic (ie, TNF blocker and/or vedolizumab) or conventional (ie, corticosteroids and/or the immunomodulators 6-mercaptopurine or azathioprine) therapy.
*In the induction study, patients were randomized to receive STELARA® weight-based dosage regimen (approximately 6 mg/kg), STELARA® 130-mg IV dose, or IV placebo. The 130-mg IV dose is not an approved induction dose and therefore is not shown.
†Clinical remisssion was defined as Mayo stool frequency subscore of 0 or 1, Mayo rectal bleeding subscore of 0, and Mayo endoscopy subscore of 0 or 1 (modified so that 1 does not include friability).
‡Weight-based induction dosage regimen: STELARA® 260 mg (weight 55 kg or less), STELARA® 390 mg (weight >55 kg to 85 kg), and STELARA® 520 mg (weight >85kg).
§The maintenance study included a third randomized arm where patients received STELARA® 90 mg subQ every 12 weeks. STELARA® 90 mg subQ every 12 weeks is not an approved maintenance dose and therefore is not shown. Patients randomized in the maintenance study were those who had a clinical response to STELARA® IV at Week 8 during the induction study.
||Week 44 of the Maintenance Study was defined as 1 year from initiation of the induction (8-week induction study + 44-week maintenance study=1 year).
¶Symptomatic remission was defined as no rectal bleeding (Mayo rectal bleeding subscore of 0) and normal to near-normal stool frequency (Mayo stool frequency subscore of 0 or 1), no endoscopic assessment was performed.
#Clinical response was defined as a decrease from baseline in the Mayo score by ≥30% and ≥2 points, with either a decrease from baseline in the rectal bleeding subscore of ≥1 or a rectal bleeding subscore of 0 or 1.
**Endoscopic improvement was defined as Mayo endoscopy subscore of 0 or 1 (modified so that 1 does not include friability).
††Patients who achieved clinical response to STELARA® at the end of the induction study.
‡‡Steroid free clinical remission was defined as patients in clinical remission and not receiving corticosteroids at 1 year.
§§Maintenance of clinical remission at 1 year was defined as remission at Week 44 in patients who achieved clinical remission 8 weeks after induction.
IIIIHEMI was defined as combined endoscopic improvement (Mayo endoscopy subscore of 0 or 1) and histologic improvement of the colon tissue (neutrophil infiltration <5% of crypts, no crypt destruction, and no erosions, ulcerations, or granulation tissue).
References: 1. STELARA® [Prescribing Information]. Horsham, PA: Janssen Biotech, Inc. 2. Data on file. Janssen Biotech, Inc.